
Long Term Follow-Up After
Administration of Human Gene
Therapy Products
Guidance for Industry
Additional copies of this guidance are available from the Office of Communication, Outreach
and Development (OCOD), 10903 New Hampshire Ave., Bldg. 71, Rm. 3128, Silver Spring,
from the Internet at https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-
regulatory-information-biologics/biologics-guidances.
For questions on the content of this guidance, contact OCOD at the phone numbers or email
address listed above.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Biologics Evaluation and Research
January 2020
Contains Nonbinding Recommendations
i
Table of Contents
I. INTRODUCTION............................................................................................................. 1
II. SCOPE ............................................................................................................................... 2
III. BACKGROUND ............................................................................................................... 2
A. Potential Risks of Delayed Adverse Events Following Exposure to Human
Gene Therapy Products ........................................................................................ 2
B. History .................................................................................................................... 3
C. Experience Gained Through Long Term Follow-up of Subjects in Gene
Therapy Trials ....................................................................................................... 4
D. Long Term Follow-up for Novel Gene Therapy Products ................................ 5
IV. PRECLINICAL DATA USED FOR ASSESSMENT OF DELAYED RISKS IN
GENE THERAPY CLINICAL TRIALS ........................................................................ 5
A. Criteria to Assess Potential Delayed Risks of Gene Therapy Products ........... 5
B. Considerations for Preclinical Study Design to Assess Biodistribution and
Persistence of Gene Therapy Product ................................................................. 9
C. Vector Persistence, Integration, and Reactivation and Genome Modification:
Assessing Long Term Risks................................................................................ 11
D. Considerations for Preclinical Evaluation of Products that Involve Genome
Editing .................................................................................................................. 15
V. RECOMMENDATIONS FOR PROTOCOLS FOR LONG TERM FOLLOW-UP
OBSERVATIONS: CLINICAL CONSIDERATIONS ............................................... 15
A. Goals of the Long Term Follow-up Observations ............................................ 15
B. Clinical Trial Populations for Long Term Follow-up Observations .............. 16
C. Duration of Long Term Follow-up Observations ............................................ 16
D. Elements of Long Term Follow-up Observations ............................................ 17
E. Informed Consent in Trials Involving Long Term Follow-up Observations 21
F. Special Considerations Regarding Integrating Vectors .................................. 22
G. Special Considerations Regarding Product Involving Genome Editing ........ 26
VI. GENERAL CONSIDERATIONS FOR POST-MARKETING MONITORING
PLANS FOR GENE THERAPY PRODUCTS ............................................................ 26
VII. LONG TERM FOLLOW-UP UNDER SPECIAL CIRCUMSTANCES .................. 27
VIII. DEFINITIONS ................................................................................................................ 28
IX. REFERENCES ................................................................................................................ 30
APPENDICES ............................................................................................................................. 33

Contains Nonbinding Recommendations
1
Long Term Follow-Up After Administration of Human Gene
Therapy Products
Guidance for Industry
This guidance represents the current thinking of the Food and Drug Administration (FDA or
Agency) on this topic. It does not establish any rights for any person and is not binding on FDA
or the public. You can use an alternative approach if it satisfies the requirements of the
applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff
responsible for this guidance as listed on the title page.
I. INTRODUCTION
We, FDA, are providing you, a sponsor who is developing a human gene therapy product (GT
Product),
1
recommendations regarding the design of long term follow-up studies (LTFU
observations) for the collection of data on delayed adverse events following administration of a
GT product. Often, GT products are designed to achieve therapeutic effect through permanent or
long-acting changes in the human body. As a result of long term exposure to an investigational
GT product, study subjects may be at increased risk of undesirable and unpredictable outcomes
that may present as delayed adverse event(s). To understand and mitigate the risk of a delayed
adverse event, subjects in gene therapy trials may be monitored for an extended period of time,
which is commonly referred to as the “long term follow-up” (LTFU) period (of a clinical study).
LTFU observations are extended assessments that continue some of the scheduled observations
of a clinical trial past the active follow-up period, and are an integral portion of the study of
some investigational GT products. LTFU observations are important to monitor long term safety
of GT products. For GT products that present long term risks to subjects, LTFU/surveillance
plan(s) should also be put in place post-licensure for monitoring of delayed adverse events (for
details we refer you to section VI. of this document). Not all GT products will require LTFU
observations; a risk assessment should be performed by a sponsor based on several factors as
outlined in this guidance.
In this guidance, we provide a brief introduction of the product characteristics, patient-related
factors, and the preclinical and clinical data that should be considered when assessing the need
for LTFU observations for your GT product. We also provide recommendations for the study
design of LTFU observations, with specific considerations for different GT products and
recommendations on patient monitoring for licensed GT products. Definitions of terms used
throughout this guidance are provided in section VIII. of this document.
This guidance finalizes the draft guidance of the same title dated July 2018 and supersedes the
document entitled “Guidance for Industry: Gene Therapy Clinical Trials – Observing Subjects
1
See section VIII. Definitions: Human gene therapy product.

Contains Nonbinding Recommendations
2
for Delayed Adverse Events” dated November 2006 (Ref. 1) (2006 Delayed Adverse Events).
This guidance is also intended to supplement the guidance entitled “Testing of Retroviral Vector-
Based Human Gene Therapy Products for Replication Competent Retrovirus during Product
Manufacture and Patient Follow-up; Guidance for Industry” dated January 2020.
2
FDA’s guidance documents, including this guidance, do not establish legally enforceable
responsibilities. Instead, guidances describe the FDA’s current thinking on a topic and should be
viewed only as recommendations, unless specific regulatory or statutory requirements are cited.
The use of the word should in FDA’s guidances means that something is suggested or
recommended, but not required.
II. SCOPE
This guidance applies to all gene therapy clinical studies and to licensed GT products for which
LTFU observations are warranted based on analyses of available preclinical and clinical safety
data for the GT product that raises concerns for delayed adverse events. The recommendations
in this guidance apply to human GT products that produce long lasting genetic effects and the
performance of LTFU observations for evidence of delayed adverse events, i.e., adverse events
that occur past the active follow-up period after exposure to the GT product, as described in the
main study protocol.
3
III. BACKGROUND
A. Potential Risks of Delayed Adverse Events Following Exposure to Human
Gene Therapy Products
Characteristics unique to human GT products that may be associated with delayed
adverse events include:
1. The integration activity of the GT product: The biological activity of
retroviral vectors
4
(e.g., vectors derived from gammaretrovirus, lentivirus,
foamy virus etc.) and transposon elements is imparted by an integration
event in the genome. In general, such integration is not directed to
specific sites in the human genome, and this raises the potential for
disruption of critical host (human) genes at the site of integration, or
activation of proto-oncogenes near the integration site(s) and, thereby, the
risk for malignancies.
2
“Testing of Retroviral Vector- Based Human Gene Therapy Products for Replication Competent Retrovirus during
Product Manufacture and Patient Follow-up; Guidance for Industry” is available at this website:
https://www.fda.gov/media/113790/download
.
3
This guidance does not apply to vaccines for infectious disease indications, bacteriophage products, live
biotherapeutic products, fecal microbiota for transplantation (FMT) products and allergenic products.
4
See section VIII. Definitions: Vector.
Contains Nonbinding Recommendations
3
2. Genome editing activity: Genome editing-based GT products impart their
biological activity through site-specific changes in the human genome, but
may also have off-target effects on the genome (Ref. 2). Similar to
integrating vectors, genome editing may produce undesirable changes in
the genome (whether ex vivo or in vivo), with the risk of malignancies,
impairment of gene function, etc.
3. Prolonged expression: A GT product where the transgene (therapeutic
gene) encodes growth factors, such as vascular endothelial growth factor
(VEGF) or proteins associated with cell division such as p53, may raise
the potential for unregulated cell growth and malignancies due to
prolonged exposure to the therapeutic protein. Similarly, transgenes
encoding immune recognition factors may introduce the risk for
autoimmune-like reactions (to self-antigens) upon prolonged exposure.
For GT products that carry transcriptional regulatory elements (e.g.,
microRNA) or immune-modulatory proteins (e.g., cytokines) there is also
the risk of unknown pleotropic effects, including altered expression of
host (human) genes that could result in unpredictable and undesirable
outcomes.
4. Latency: When the GT product has the potential for latency, such as a
herpesvirus, there is the potential for reactivation from latency and the risk
of delayed adverse events related to a symptomatic infection.
5. Establishment of persistent infections: GT products that are replication
competent viruses and bacteria, such as listeria-based bacterial vectors,
have the potential to establish persistent infections in
immunocompromised patients leading to the risk of developing a delayed
but serious infection.
In addition to product-related factors, the long term risk profile of a GT product should
also take into consideration the target cell/tissues/organ, and the patient population (age,
immune status, risk of mortality etc.), and the relevant disease characteristics.
B. History
The recommendations for LTFU monitoring in the 2006 Delayed Adverse Events
guidance (Ref. 1) were based on extensive discussions among gene therapy stakeholders,
and cumulative preclinical and clinical experience with GT products (Refs. 3, 4, 5) as
summarized in this section. To discuss and solicit advice about long term risks to
subjects exposed to such products, three separate meetings of the FDA advisory
committee, Biological Response Modifiers Advisory Committee (BRMAC), were
convened on November 17, 2000, April 6, 2001, and October 24, 2001 (Ref. 6).
A public workshop entitled “Long-term Follow-Up of Participants in Human Gene
Transfer Research” was also held in June 2001, in association with the annual meeting of
Contains Nonbinding Recommendations
4
the American Society of Gene Therapy (ASGT). The workshop included a forum in
which invited speakers discussed the challenges associated with LTFU of subjects in
gene therapy clinical studies. The workshop organizers published a summary of the
discussion (Ref. 7).
Taking these discussions into consideration, we provided detailed recommendations in
the 2006 Delayed Adverse Events guidance document on the duration and design of
LTFU observations (Ref. 1). The Agency advised sponsors to observe subjects for
delayed adverse events for as long as 15 years following exposure to the investigational
GT product, specifying that the LTFU observation should include a minimum of five
years of annual examinations, followed by ten years of annual queries of study subjects,
either in person or by questionnaire.
Herein, we update our recommendations in the guidance taking into account the clinical
experience gained since 2006 in LTFU of investigational GT products (Refs. 8, 9 and as
described in the following section), and the development of novel GT products with
emerging technologies such as genome-editing that may be associated with an increased
risk of delayed adverse events (as described in section III.D of this document).
C. Experience Gained Through Long Term Follow-up of Subjects in Gene
Therapy Trials
To date, leukemias have been reported in more than one trial where subjects have
received genetically-modified cells that were manufactured using gammaretroviral
vectors (Refs. 10-13). Advances in analytical approaches for integration site analysis in
patient samples collected during LTFU have provided insight into the possible
mechanisms involved in the occurrence of such delayed adverse events (Refs. 10-16).
Past clinical experience in LTFU monitoring, and significant improvements in analytical
approaches to investigate the integration site have contributed greatly towards our
understanding of the risks associated with integrating gene therapy vectors (Ref. 17).
Such risks can be mitigated through improvements in vector design and the duration and
design of LTFU observations. Because integrating gene therapy vectors can persist in the
body over the life-span of the patient’s transduced cells, vectors with an improved risk
profile were desired, and have subsequently been developed for clinical use (Refs. 18,
19). These include gammaretroviral and lentiviral vectors modified:
1. To reduce the risk of activating host genes adjacent to the integration site
(e.g., self-inactivating (SIN) vectors and vectors containing insulator
sequences);
2. To be less genotoxic (e.g., carrying non-viral physiological promoters to
drive the expression of the therapeutic gene); and
3. To reduce the potential for recombination, and thereby, the risk of
generating replication competent, pathogenic variants.

Contains Nonbinding Recommendations
5
D. Long Term Follow-up for Novel Gene Therapy Products
Novel GT products developed as a result of emerging technologies, such as transposon-
based gene insertion and genome editing, also raise concerns for delayed adverse events
due to the unique genome modifying activity of such products. Specifically, a vector
with a transposon element can insert transgenes into the host chromosome randomly by a
direct “cut-and-paste” mechanism, mediated by the transposases (enzyme) activity in the
product (Ref. 20). A GT product with genome editing components can give rise to non-
specific off-target changes in the genome (Ref. 2), and may be associated with unknown
and unpredictable risks for developing delayed adverse events in study subjects and
patients (once approved), the extent of which will vary depending on the targeting
mechanisms accompanying these components. The LTFU observations for these novel
GT products should be designed to take into account product-specific characteristics, the
basic and translational knowledge generated in the field, and the product-specific
preclinical data generated to enable investigational new drug application (IND) studies,
as described in the following section.
IV. PRECLINICAL DATA USED FOR ASSESSMENT OF DELAYED RISKS IN
GENE THERAPY CLINICAL TRIALS
A. Framework to Assess Potential Delayed Risks of Gene Therapy Products
To assess the risk of delayed adverse events for a GT product, we recommend that you
use available preclinical and clinical evidence, and current information about your
product and similar products based on studies that you and others have performed. In
general, when the risk of delayed adverse events is low following exposure to a GT
product, LTFU observations are not recommended. We consider the assessment of risk
to be a continuous process; as more data accumulates, we recommend that you reassess
the risk to your subjects and, if appropriate, revise your existing LTFU observations
study protocol or initiate a LTFU observations study protocol, if previously allowed to
proceed without an LTFU observations study protocol.
Preclinical and clinical experience with your product or similar products may be
considered relevant in the assessment of the risk for delayed adverse events. For
example, experience with GT products in the same vector type
5
, administered by a
similar route, or given for the same clinical indication may contribute helpful
information. However, for novel products such information may not be available or
applicable, or may be limited, in which case data from well-designed preclinical studies
(as described in section IV.B of this document) should be used in assessing the risk of
delayed adverse events. Primary data and information relevant to the assessment of the
risk of delayed events should be submitted in your IND along with other preclinical data
(see 21 CFR 312.23(a)(8), 312.23(a)(10)(iv), and 312.23(a)(11)).
5
See Table 1 for examples of vector types.
Contains Nonbinding Recommendations
6
GT product knowledge is critical in assessing the level of risk for delayed adverse events
and the need for LTFU observations. To help you in this process, we refer you to section
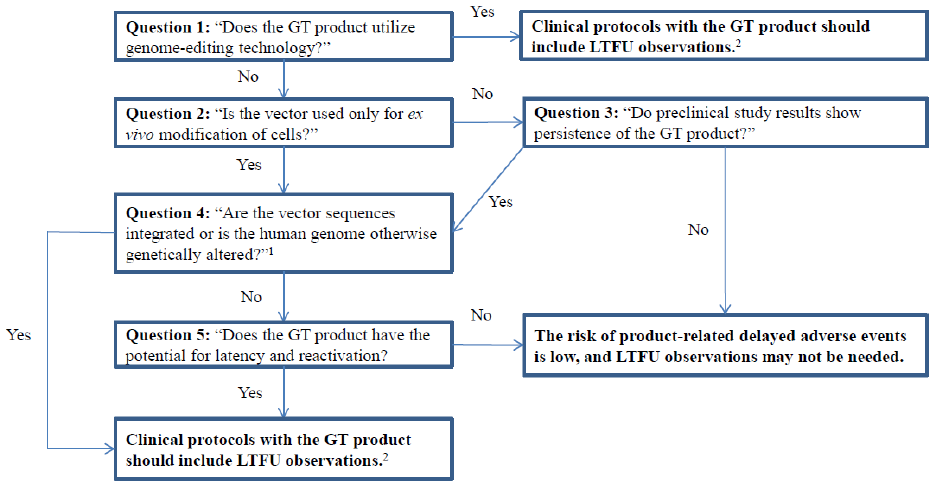
III.A of this document, and to the series of questions in Figure 1, “Framework to Assess
the Risk of Gene Therapy-Related Delayed Adverse Events.”
Figure 1. Framework to Assess the Risk of Gene Therapy-Related Delayed Adverse Events
1
If you have evidence that suggests that the product may integrate or if the product was intentionally
designed to facilitate integration (please refer to Table 1, section IV.C of this document); the answer is
“yes.”
2
See section V. of the text for recommendations on how to perform clinical LTFU observations.
Note, that evidence from preclinical studies will help you answer questions 3 through 5
below and in Figure 1. When the risk of delayed adverse events is low based on your
answers to these questions, a plan for LTFU observations may not be necessary to
mitigate risks to subjects.
We suggest you use the framework in Figure 1 by answering the questions in sequence as
follows:
Question 1: “Does your GT product utilize genome-editing technology?”
If the answer is “no,” go to Question 2. If the answer is “yes,” all your clinical
protocols proposing administration of the GT product should include LTFU
observations for appropriate human subject protections (see section V. for
recommendations on how to perform clinical LTFU observations).
Question 2: “Is your vector used only for ex vivo modification of cells?”

Contains Nonbinding Recommendations
7
If the answer is “no,” go to Question 3. If the answer is “yes,” go to Question 4.
Question 3: “Do preclinical study results show persistence of the GT product?”
If the answer is “no,” the risk of product-related delayed adverse events is low,
and LTFU observations may not be needed. If the answer is “yes,” go to
Question 4.
If it is unknown whether your GT product persists, for the purpose of assessing
the risk of delayed adverse events, we recommend that you either assume that the
GT product does persist, or perform preclinical studies to assay for the GT
product persistence in a relevant animal species (as described in section IV.B of
this document and Ref. 21). For recommendations concerning the design and
details of such preclinical studies, please refer to section IV.B of this document.
Specifically, we recommend the polymerase chain reaction (PCR) assay for
determining vector persistence in biodistribution studies. Following
administration of the product, persistence is indicated by detectable levels of GT
product sequences above the threshold level (<50 copies/μg genomic DNA) of
the PCR assay, and absence of an apparent downward trend over several time
points. In contrast, persistence is unlikely if product sequences cannot be detected
with a sensitive assay such as PCR or if the assay for GT product sequences
demonstrates a downward trend over time. We encourage you to consult with the
Office of Tissues and Advanced Therapies (OTAT) at the Center for Biologics
Evaluation and Research (CBER) for specific advice regarding determination of
GT product persistence and biodistribution in your test system.
Question 4: “Are your vector sequences integrated or is the human genome
otherwise genetically altered?”
If the answer is “no,” go to Question 5. If you have evidence that suggests that
the product may integrate or if the product was intentionally designed to facilitate
integration (please refer to Table 1, section IV.C of this document); the answer is
“yes.” If the answer is “yes,” all your clinical protocols proposing administration
of the GT product should include LTFU observations for appropriate human
subject protections (see section V. for recommendations on how to perform
clinical LTFU observations).
Question 5: “Does the GT product have the potential for latency and
reactivation?”
If the answer is “no,” the risk of product-related delayed adverse events is low,
and LTFU observations may not be needed. If the answer is “yes,” all your
clinical protocols with the GT product should include LTFU observations for
appropriate human subject protections (see section V. for recommendations on
how to perform clinical LTFU observations).
Contains Nonbinding Recommendations
8
Laboratory and preclinical evidence of a low risk of delayed adverse events following
exposure to a similar GT product may show that LTFU observations for your GT product
are not needed. When such data/information is made available for review, we can assess
their relevance to your product if you provide adequate details and a clear explanation of
similarities and differences between the two products. For additional guidance, we
provide the following two examples:
• Your GT product is a plasmid, and the similar product is also a plasmid,
but has different coding sequences for the proposed therapeutic gene
product. The similar product has been used in preclinical and clinical
studies, administered by an identical route and in an identical final
formulation to that proposed in the prospective studies in your program. In
this case, reference to a published study demonstrating lack of persistence
of the vector sequence for the similar (plasmid) product may adequately
address concerns regarding the persistence of the proposed vector (your
plasmid).
• Your GT product and the similar product differ only with respect to route
of administration. The similar product was administered into tumors
(intratumorally). Your GT product is to be administered intravenously.
There is a published study demonstrating the lack of persistence of the
similar product when administered intratumorally. In this case, the data is
not sufficiently relevant to the GT product under study, since there was no
intended systemic exposure to the product. Thus, there is insufficient
similarity to conclude that LTFU observations are not warranted in your
proposed study to mitigate the long term risks to subjects. In the absence
of relevant data from a study involving a similar product, we recommend
that you assess the risk of product persistence in a preclinical study with
the proposed GT product administered by the intravenous route.
If you believe you have evidence from studies on a similar product that is adequate to
support conclusions that either the GT product is unlikely to persist in human hosts, or
the vector sequence does not integrate into the human genome and the GT product does
not have the potential for latency and reactivation, you may decide to submit a clinical
protocol that does not provide for LTFU observations. We will review such submissions
and, if based upon our review of your submission or other additional information, we
conclude that LTFU observations for delayed adverse events are necessary to mitigate
long term risks, and that without LTFU observations, the study presents an unreasonable
and significant risk to study subjects, we may place your study on clinical hold (21 CFR
312.42(b)(1)(i) and 312.42(b)(2)(i)).
We provide the following examples of evidence obtained from investigation of a product
that may warrant our recommendation of LTFU observations for delayed adverse events:
• A preclinical toxicology study indicates that expression of the therapeutic
gene (the transgene in your product) is associated with delayed toxicity.

Contains Nonbinding Recommendations
9
• The therapeutic gene provides functional replacement of a host gene that
is otherwise not expressed, and the therapeutic protein is potentially
immunogenic.
• Data collected in a clinical study with your GT product indicates product
persistence, even though data from your preclinical studies suggested that
the product did not persist.
• Data collected in a clinical study with your GT product identifies an
increased risk of delayed adverse events.
B. Considerations for Preclinical Study Design to Assess Biodistribution and
Persistence of Gene Therapy Product
As discussed in section III.A of this document, product persistence heightens the risk of
delayed adverse events following exposure to the GT product. Indeed, the longer the GT
product persists, the greater the duration and degree of risk of delayed adverse events.
We recommend that you perform preclinical biodistribution studies using methods shown
to be sensitive and quantitative to detect product sequences. Such studies would be
designed to determine the distribution of your product in non-target tissues and the
persistence of the product in both non-target and target tissues following direct in vivo
administration of the product.
6
If possible and applicable, we recommend that the studies
employ an animal species that permits vector transduction and/or vector replication and
that the animal species be biologically responsive to the specific transgene of interest or
to therapeutic components in the product (e.g., for products that may not contain
transgenes and only genome editing components) (Ref. 21). The duration of the
preclinical studies will vary, depending on the animal model employed. Projections of
delayed adverse reactions in human subjects may be derived from assessment of data
from appropriate long term observational studies in animals, when such observational
studies are possible.
A biodistribution study in animals can be performed either as a separate study or as a
component of a pharmacology or toxicology study.
7
We recommend that you consider
the following points in your animal study design to permit evaluation of GT product
localization and persistence (Ref. 22).
6
In contrast to biodistribution assessment, ‘shedding’ describes how the vector product is excreted or released from
the patient’s body (Guidance for Industry: Design and Analysis of Shedding Studies for Virus or Bacteria-Based
Gene Therapy and Oncolytic Products; August 2015 available at https://www.fda.gov/media/89036/download ).
7
For additional information on this topic refer to the 2018 International Pharmaceutical Regulators Programme
(IPRP) reflection paper, titled, “Expectations for Biodistribution (BD) Assessments for Gene Therapy (GT)
Products” (
http://development.iprp.backend.dev6.penceo.com/sites/default/files/2018-
09/IPRP_GTWG_ReflectionPaper_BD_Final_2018_0713.pdf).
Contains Nonbinding Recommendations
10
1. Animal Study Design
a. Use the GT product in the final formulation for the clinical study
because changes in the final formulation may alter biodistribution
pattern.
b. Use both sexes or justify the use of a single sex.
c. Use at least 5 animals per sex per group per sacrifice time point for
rodents, and between 3-5 animals per sex per group per sacrifice
time point for non-rodents.
d. Consider factors in the study design that might affect the GT
product distribution and/or persistence such as the animal’s age
and physiologic condition.
e. Use the intended clinical route of GT product administration, if
possible.
f. Assess GT product biodistribution in a vehicle control group and a
group of animals that receives the maximum proposed clinical
dose level. Studies at additional dose levels might provide
information on dose-dependent effects of your product.
g. Inclusion of appropriate safety endpoints in your biodistribution
study may be helpful to assess any potential correlation between
product presence/persistence and adverse findings . These
endpoints can include clinical observations, body weights, clinical
pathology, gross organ pathology, and histopathology.
h. Include several sacrifice intervals to characterize the kinetics of
GT product distribution and persistence. We recommend sacrifice
of animals at the expected time of peak GT product detection and
at several later time points to evaluate clearance of product
sequences from tissues.
2. Tissue Collection and Analysis
a. Sample and analyze the following panel of tissues, at a minimum:
blood, injection site(s), gonads, brain, liver, kidneys, lung, heart,
and spleen. Consider other tissues for evaluation, depending on
the product, vector type and tropism, and transgene(s), as well as
the route of administration (e.g., draining lymph nodes and
contralateral sites for subcutaneous/intramuscular injection, bone
marrow, eyes, etc.).
b. Choose a method for tissue collection that avoids the potential for
cross contamination among different tissue samples (i.e., in order
of least to highest expected presence of vector).
c. Use a quantitative, sensitive assay such as quantitative PCR
(qPCR), to analyze the samples for vector sequences. You should
submit data to your IND to demonstrate that your assay
methodology is capable of specifically detecting vector sequence
in both animal and human tissues. We recognize that analytical

Contains Nonbinding Recommendations
11
technologies are constantly changing, and encourage you to
discuss the assay methodology with us before initiating sample
analysis. Our current qPCR recommendations include the
following:
i. The assay should have a demonstrated limit of quantitation
of <50 copies/μg genomic DNA, so that your assay can
detect this limit with 95% confidence.
ii. The DNA samples should be run in triplicate for each
tissue. To aid the interpretation of the qPCR assay results,
one replicate of each tissue sample should include a spike
of control DNA, including a known amount of the vector
sequences. The spike control will determine the specified
qPCR assay sensitivity.
iv. In the final study report, individual animal data should be
provided. The method for how values below the Limit of
Quantitation of the assay are categorized and calculation of
the median or mean value should be specified.
3. Other Considerations
There are many variables that will affect the outcome and interpretation of
the in vivo assessment of each GT product type. Hence, we encourage
you to discuss with OTAT the study design for your GT product in a pre-
IND meeting before initiating the preclinical biodistribution study to
ensure that both biodistribution and persistence will be adequately
assessed.
8
C. Vector Persistence, Integration, and Reactivation and Genome Modification:
Assessing Long Term Risks
GT products may or may not use technologies that modify the host genome. For products
that do, such as integrating vectors (gammaretrovirus, lentivirus, foamy virus etc.),
herpesvirus capable of latency-reactivation, and genome editing products (as described
under sections III.A and III.D of this document, respectively), there is the risk of delayed
adverse events. Accordingly, as depicted in Table 1 of this document and in the answer
to Question 4 in Figure 1, it is important to conduct LTFU observations to mitigate
delayed risks to subjects receiving GT products with integrating activity.
8
The preclinical program for any investigational product should be individualized with respect to scope, complexity,
and overall design, to maximize the contribution and predictive value of the resulting data for clinical safety and
therapeutic activity. We encourage sponsors to explore opportunities for reducing, refining, and replacing animal
use in the preclinical program. For example, it may be appropriate to use in vitro or in silico testing to complement
or replace animal studies. Sponsors are encouraged to submit proposals and justify any potential alternative
approaches, which we will evaluate for equivalency to animal studies.
Contains Nonbinding Recommendations
12
We are aware that the potential of vectors to integrate may be modified to increase their
utility as gene therapy agents; for example, a vector can be modified to induce integration
of its DNA (Refs. 23-26). Another example would be changes in the methods used to
introduce plasmid DNA vectors into cells that result in higher integration frequencies
(Ref. 27). In those cases where a modification of the GT product may have altered its
persistence or integration properties, we recommend that you submit data to your IND
from preclinical studies to assess vector persistence in an appropriate model and take one
of the following actions:
1. If the vector is not persistent, the predicted risk of delayed adverse events
would appear to be low in which case LTFU observations may not be
needed.
2. If the vector is persistent, we recommend that you perform preclinical
studies to assess vector integration, as well as the potential for vector
latency and reactivation.
3. If the studies show no evidence for persistence due to integration of the
genetic material or development of latency, the predicted risk of delayed
adverse events would be low. LTFU observations may not be needed.
4. If the studies show no evidence for integration of the genetic material but
studies for latency and reactivation are inconclusive, cannot be performed,
or show evidence of latency and/or reactivation, the predicted risk of
delayed adverse events is indeterminate. LTFU observations may be
recommended for human subject protections.
5. If preclinical studies of vector integration are not feasible, if the
therapeutic gene/genetic material integrates, or if the vector is shown to
persist in a latent state that may be reactivated, the risk of delayed adverse
events is high or unknown, then LTFU observations in study subjects are
recommended for human subject protection.
6. If vector integration studies are not performed, we recommend that you
provide other evidence to support an assessment that your product does
not pose high risks of delayed adverse events, including the following:
a. A discussion of why vector integration studies were not performed.
b. The evidence supporting your assessment of the risk of delayed
adverse events posed by your product.
As stated in section IV.B.3 of this document, we encourage you to discuss with FDA
your study design before starting the trial.
GT products that are based on vectors such as plasmids, poxvirus, adenovirus, and adeno-
associated virus vectors (AAV) that do not have a propensity to integrate or reactivate
Contains Nonbinding Recommendations
13
following latency, generally present a lower risk of delayed adverse events. Clinical data
from LTFU observations of subjects that have received plasmids, poxvirus, adenovirus,
and AAV in trials conducted since 2006, further supports the assessment of lower risk for
these GT products. However, vector or product-specific modifications may alter the risk
profile of products that are currently considered lower risk, for example a plasmid that is
modified to carry genome editing components. Conversely, gene therapy vectors
currently considered to pose delayed risks might be modified in order to reduce those
risks. Hence, data supporting decreased or increased risk for delayed adverse events with
novel GT products or vector types could provide the basis for sponsors to reassess our
recommendations for performing LTFU observations. We encourage you to consult with
OTAT regarding a reassessment of our recommendations for performing LTFU
observations.
Contains Nonbinding Recommendations
14
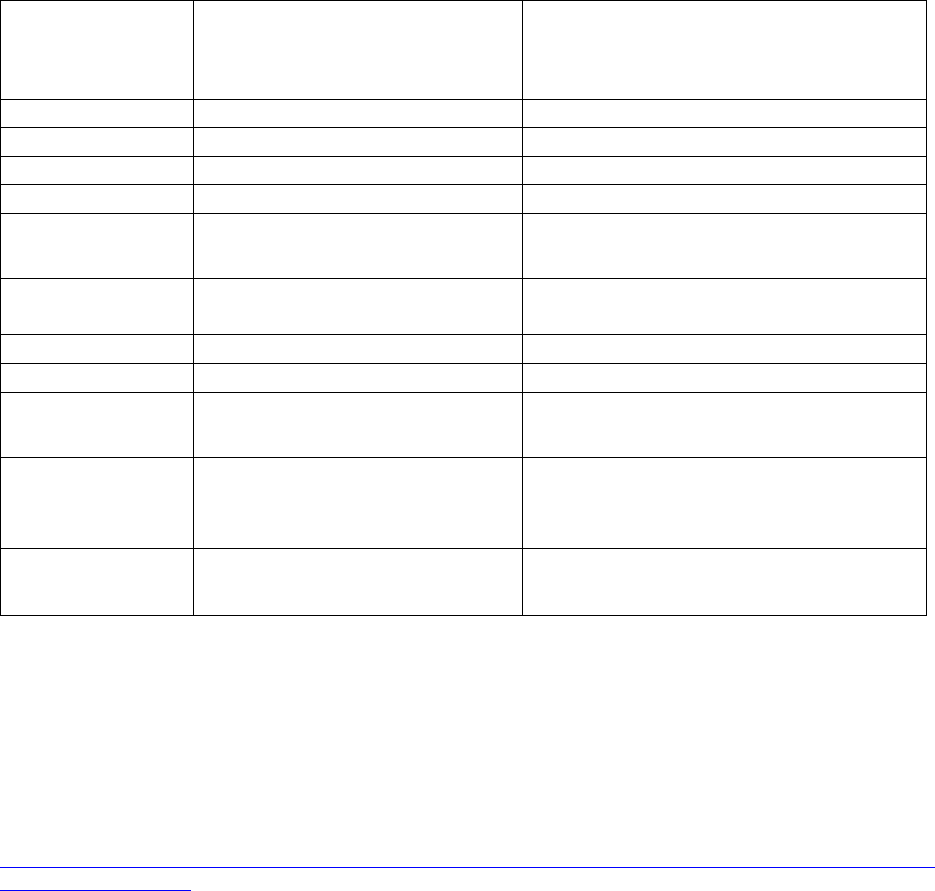
Table 1. Propensity of Commonly Used Gene Therapy Products/Vectors to Modify the
Host Genome
Product/Vector
Type
Propensity to Modify Genome
1
Long Term Follow-up Observations
2
Plasmid
No
No
RNA
No
No
Poxvirus
No
No
Adenovirus
No
No
Adeno-
associated virus
3
No Product specific
Herpesvirus
No, but may undergo
latency/reactivation
Yes
Gammaretrovirus
Yes
Yes
Lentivirus
Yes
Yes
Transposon
elements
Yes Product specific
Microbial vectors
for gene therapy
(MVGT)
4
No, but may persist and undergo
reactivation
Product specific
Genome editing
products
Yes; permanent changes to the
host genome
Yes
1
Based on product design (i.e., lack of any known mechanism to facilitate integration or genome editing), as well as
cumulative preclinical and clinical evidence suggesting that a GT product does not integrate into or edit the genome
or integrates in/modifies the genome at very low frequencies.
2
Specific circumstances that indicate persistent expression of the transgene, in the absence of integration or genome
editing, may be the basis for a conclusion that LTFU observations are recommended to mitigate long term risks to
subjects receiving these vectors. This would depend on additional criteria, such as the transgene expressed or
clinical indication, as described in this section.
3
Replication-negative vectors only.
4
For additional guidance we refer you to “Recommendations for Microbial Vectors used for Gene Therapy;
Guidance for Industry” dated September 2016,
https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandG
eneTherapy/default.htm.
Contains Nonbinding Recommendations
15
D. Considerations for Preclinical Evaluation of Products that Involve Genome
Editing
Genome editing, whether ex vivo or in vivo, introduces the risk for delayed adverse
effects, due to 1) the permanent nature of change; 2) the potential for off-target genome
modifications that can lead to aberrant gene expression, chromosomal translocation,
induce malignancies, etc.; 3) the risk for insertional mutagenesis when integrating vectors
are used to deliver the genome editing components, and the associated risk of
tumorigenicity; and/or 4) the possibility of an immune response to the genome-editing
components or the expressed transgene. Preclinical safety evaluation of genome editing
products should consider: 1) the technology used to edit the genome; 2) the target cell
types that are modified; 3) the genomic sites that are modified, 4) the vector used to
deliver the genome-editing components; and 5) the clinical route of administration.
Preclinical studies evaluating these factors can inform the scope of the clinical LTFU
observations.
For guidance on the biodistribution studies when considering the vector type in the
genome edited product, and the related long term risks with integrating vectors, we refer
you to sections IV.B and IV.C of this document.
V. RECOMMENDATIONS FOR PROTOCOLS FOR LONG TERM FOLLOW-UP
OBSERVATIONS: CLINICAL CONSIDERATIONS
In this section, we recommend elements appropriate to the design and conduct of LTFU
observations for delayed adverse events in study subjects receiving investigational GT products.
Typically, LTFU observations are conducted under a protocol (LTFU protocol) that is separate
from the main study protocol or as an extension of the main protocol study, and may begin
immediately after the first subject completes their last visit in the main study and enrolls in the
LTFU study.
A. Goals of the Long Term Follow-up Observations
The objective of LTFU observations in clinical development of a GT product is to
identify and mitigate the long term risks to the patients receiving the GT product. The
LTFU protocol for gene therapy trials is primarily designed to capture delayed adverse
events in study subjects as well as to understand the persistence of the GT product. As a
sponsor, you may consider designing the LTFU protocol to assess the long term clinical
efficacy, and durability of your product. For additional guidance on trial design for GT
products we refer you to FDA’s guidance document “Considerations for the Design of
Early-Phase Clinical Trials of Cellular and Gene Therapy Products; Guidance for
Industry” dated August 2015 (Ref. 28).
Contains Nonbinding Recommendations
16
B. Clinical Trial Populations for Long Term Follow-up Observations
When a GT product is deemed to pose a risk for delayed adverse events (based on the
recommendations/discussions provided under sections III and IV of this document) and a
decision to perform LTFU observations is made, all study subjects who receive the GT
product are expected to be enrolled in the LTFU protocol (as discussed in Section V.C of
this guidance) after signing an informed consent document. LTFU observations may
have reduced utility in assessing and mitigating subject risk when the population selected
for the trial has characteristics that could confound the observation of the delayed adverse
events, such as short life expectancy, multiple co-morbidities, and exposure to other
agents such as radiation or chemotherapy. In contrast, LTFU observations could have
greater value in assessing and mitigating the risks to subjects who have limited disease or
are disease-free, and who have few co-morbidities and limited exposures to other agents
with potential for delayed adverse events. Hence, characteristics of the patient population
and the disease to be treated should be considered when designing a LTFU protocol.
C. Duration of Long Term Follow-up Observations
It is important that the design of LTFU observations be appropriate to detect potential
gene therapy-related delayed adverse events in the subjects enrolled in your clinical
studies. The duration of LTFU should be sufficient to observe the subjects for risks that
may be due to the characteristics of the product, the nature of the exposure (e.g. route of
administration and the biodistribution profile), and the anticipated time of occurrence of
delayed adverse events. Elements that will influence the determination of the duration of
LTFU observations include the following:
• The observed duration of in vivo product persistence.
• The observed duration of transgene expression.
• Product characteristics in vivo.
• Route of administration.
• The expected survival rates and the known background rates of the events
of interest occurring in the study population.
• Other factors that may be relevant to the feasibility and scientific value of
conducting LTFU observations; for example, the durability of the clinical
effect.
In general, our current recommendations for the duration of a LTFU protocol based on
product type are as follows:
• Fifteen years for integrating vectors such as gammaretroviral and lentiviral
vectors and transposon elements.
• Up to fifteen years for herpes virus vectors (or oncolytics) that are capable
of establishing latency.
• Up to fifteen years for microbial vectors that are known to establish
persistent infection.
Contains Nonbinding Recommendations
17
• Up to fifteen years for genome editing products.
• Up to five years for AAV vectors.
Additionally, a risk-based approach for determining the duration of a LTFU protocol may
be considered for vectors capable of latency (e.g., Herpesvirus), long term expression
without integration (e.g., AAV), or vectors carrying gene editing components.
Although these recommendations are broadly based on GT product type, you should also
consider the elements listed above, in this section, as it applies to your GT product,
disease characteristics, and the patient population, in addition to the discussions in
sections III. and IV. of this document.
To reduce the unnecessary burden to study subjects and to you as the study sponsor, it
may be appropriate to modify the duration of the LTFU observation based on your
ongoing assessment of product persistence, transgene expression, and clinical findings. If
you intend to modify the duration of the follow-up, you may submit an amendment to
your IND justifying the change to your LTFU protocol, and communicate with FDA to
reach a final decision (we refer you to section V. of this document for additional guidance
regarding amendments to the clinical protocol).
FDA recommends sponsors make every effort to prevent patient loss to follow-up to the
extent feasible for completion of LTFU observations.
D. Elements of Long Term Follow-up Observations
We recommend that at least the following general elements be part of the LTFU protocol:
• You should establish a dedicated clinical LTFU protocol detailing patient
visit schedules, sampling plan (for patient test samples, such as blood),
methods of monitoring tests, and clinical events of interest that will be
monitored over the entire LTFU observation.
• The investigator is required to prepare and maintain adequate and accurate
case histories that record all observations and other data pertinent to the
investigation on each subject administered the investigational drug or
employed as a control in the investigation (see 21 CFR 312.62(b)). These
records would include a baseline history prior to exposure to the
investigational product in which all diseases, conditions and physical
abnormalities are recorded. A template for health care providers (HCPs)
who are not investigators or sub-investigators (for example, the subject’s
physician, physician assistant, or nurse practitioner) to use in recording
and reporting such observations to the investigator may be helpful for such
HCPs. Case histories should also include information from scheduled
visits with a HCP and test results for persistent vector sequences. The use
of surrogate tests may be necessary to indicate vector persistence if direct
sequence testing involves an invasive procedure for the subject. If
Contains Nonbinding Recommendations
18
surrogate tests are considered, we recommend that you consult with FDA
regarding the types and characteristics of the surrogate tests you intend to
use before including them in your study.
In addition, for the first five years or more (as applicable to your product), we
recommend that you do the following:
• Ensure that investigators maintain, in the case history, a detailed record of
exposures to mutagenic agents and other medicinal products, and have
ready access to information about their adverse event profiles.
• Establish a method for investigators to record the emergence of new
clinical conditions, such as:
- New malignancy(ies)
- New incidence or exacerbation of a pre-existing neurologic
disorder
- New incidence or exacerbation of a prior rheumatologic or other
autoimmune disorder
- New incidence of a hematologic disorder.
- New incidence of infection (potentially product-related)
• Design a plan for scheduled visits with an HCP to elicit and record new
findings for each study subject, including history, physical examination, or
laboratory testing, as applicable to the study population.
- Such a plan needs to facilitate reporting of delayed adverse events,
including unexpected illness and hospitalization by study subjects
and HCPs.
For the subsequent period of LTFU (applicable to products for which such length LTFU
is needed), at a minimum, we recommend that you ensure that your investigators:
• Contact subjects at a minimum of once a year. At your discretion, unless
the LTFU protocol provides for additional specific screening, you may
arrange to contact subjects by telephone or written questionnaire rather
than by office visits with an HCP.
• Continue appropriate follow-up methods as indicated by previous test
results. For example, it would be appropriate to monitor for vector
sequences in subjects who had previous test results demonstrating vector
persistence.
Perform all LTFU observations according to FDA regulations governing clinical trials
(Ref. 29).
We provide additional specific recommendations and requirements for data collection,
recording, and reporting of adverse events for LTFU observations as follows:

Contains Nonbinding Recommendations
19
1. Detection of Adverse Events and Coordination of Data Collection
9
a. To facilitate detection of delayed adverse events, we recommend
that the LTFU protocol identify suitable HCPs (e.g., subject’s
primary healthcare provider) whose observations would be used in
the assessment of the occurrence of adverse events in the study
population. Suitable HCPs might include physicians, physician’s
assistants, and nurse practitioners who were not otherwise
associated with the clinical trial. You may arrange to have such
individuals notified to provide prompt reports of adverse events to
the investigators. However, the investigators are responsible for
reporting appropriate adverse events to the sponsor. (21 CFR
312.64(b)).
b. To increase subject compliance and improve the quality of data
collection, we suggest that you encourage study subjects to be
proactive in reporting adverse events. Tools that study subjects
could use to report events to the investigator include subject diaries
of health-related events, informational brochures, and laminated,
wallet-sized cards with investigator contact information.
c. To determine the causality of potential related adverse events (such
as tumor formation) associated with your GT product, you should
propose a clinical program for follow-up procedures. Such a
program would lay out the efforts that would be needed among the
study subjects, HCPs, investigators, and the sponsor for study
coordination. This includes the collection of tissue samples for
follow-up analysis, obtaining informed consent for a biopsy or
autopsy (see section V.E. of this document), communicating with
the study subject, and preserving and analyzing the tissues/samples
according to the LTFU protocol. You may propose specific tests
to enable causality analyses such as general blood work,
cytogenetic and histological analysis, PCR, HLA typing, or deep
sequencing.
2. IND Safety Reports
You must follow applicable reporting requirements outlined in 21 CFR
312.32 for adverse events associated with the use of the investigational
product. As the LTFU observations proceed, you must notify FDA and
each participating investigator of any serious and unexpected suspected
adverse reaction (21 CFR 312.32(c)(1)(i)), and findings from other studies
(21 CFR 312.32(c)(1)(ii)). In each IND Safety Report (required to be
9
To improve the detection of adverse events as well as improve the coordination of data collection for LTFU, any
follow up procedures involving study subjects who are children and/or otherwise unable to communicate, should
include the study subject’s legally authorized representative, e.g. guardians, as defined in 21 CFR 50.3.
Contains Nonbinding Recommendations
20
provided to investigators and FDA), you must identify all safety reports
previously filed concerning a similar adverse finding, and analyze the
significance of the adverse finding in light of the previous, similar reports
(21 CFR 312.32(c)(1)). You must promptly investigate all safety
information you receive (21 CFR 312.32(d)(1)). If the relationship of the
adverse event to the GT product is uncertain, additional investigations
may be needed. You must also revise your informed consent document
and Investigator Brochure to include the new adverse event(s) that may be
associated with the product or study procedures (21 CFR Part 50, 21 CFR
312.55(b)). You must inform all clinical investigators of the newly
identified risk (21 CFR 312.32(c)(1)).
3. Annual Reports to the IND/Summary Information
While the IND is in effect and LTFU observations are ongoing, you must
file an annual report (21 CFR 312.33). It is recommended that the annual
report contain an independent section with a subtitle for Long Term
Follow-Up (See Appendix 1 of this document). In that report, you should
submit information obtained during the previous year's clinical and
nonclinical investigations, including, a summary of all IND safety reports
submitted during the past year, and a narrative or tabular summary
showing the most frequent and most serious adverse experiences by body
system (21 CFR 312.33(b)(1) and (2)). If adverse reactions are reported
and determined to be related to your product or delivery procedure, you
should provide causal analyses based on evidence from clinical,
laboratory, molecular, cytogenetic, histological, or HLA analysis, or deep
sequencing data. Please refer to Appendix 2 of this document for the
LTFU Annual Report Template. You may reference existing IND or
Development Safety Update Report (DSUR) subsections (e.g. preclinical
and clinical updates) for supporting information. In lieu of annual reports,
you may submit aDSUR. In this case, you should provide the LTFU
information in a subsection with a subtitle for Long-Term Follow-Up in
your DSUR report (Ref. 30).
4. Amendments to the Clinical Protocol
If clinical data suggest that your GT product is not associated with delayed
risks or there is no evidence of vector persistence, you may want to
consider revising the clinical protocol regarding LTFU of study subjects.
However, before implementation of this change, we recommend that you
consult with FDA and provide your rationale with supporting clinical and
laboratory data (we refer you to section V.C of this document for
additional guidance). You must submit to FDA a protocol amendment to
your IND indicating the relevant changes (21 CFR 312.30(b)(1), (d), and
(e)).
Contains Nonbinding Recommendations
21
5. Scheduled Physical Examinations
We recommend that LTFU observations include scheduled physical
examinations performed by a HCP once a year during the first five years
(or until the completion of LTFU if the LTFU is less than five years),
unless the assessed risks associated with your GT product indicate that
they should be done more frequently. For example, if a subject exposed to
your GT product develops a rapidly progressive, potentially reversible
delayed adverse event, and there is a reasonable possibility that the event
may have been caused by the product, it may then become advisable to
perform observations on a semi-annual or quarterly basis, or more
frequently as clinically indicated. Such periodic evaluation should include
a brief history and focused examination designed to determine whether
there is any evidence of emergence of clinically important adverse events.
Appropriate laboratory evaluations, such as a hematology profile, should
be included with the periodic physical examination. LTFU observations
are intended to collect data on delayed adverse events related to the GT
product, and are not intended to provide evaluation or treatment data for
the underlying disease.
6. GT Product Persistence
During LTFU observations, we recommend that you test study subjects at
least annually for persistent vector sequences until they become
undetectable. More frequent testing may be necessary as outlined in
section V.G of this document. The assay should be sufficiently sensitive
to detect vector sequences. We recommend that you sample the likely
population of transduced cells without being overly invasive (e.g.,
peripheral blood is a suitable sample to test for presence of hematopoietic
stem cells, rather than bone marrow biopsy). In those cases where
collecting the transduced cell population may involve an invasive
procedure, we recommend that you consider, instead, measuring a
surrogate that may indicate vector persistence (e.g., the level of transgene
product or some clinical effect). Data demonstrating the lack of detectable
vector may provide a rationale to revise the LTFU protocol as a protocol
amendment to your IND. In any such protocol amendment, include an
assessment of risks associated with your GT product and an evaluation of
the impact of the waning persistence of the vector on those risks (21 CFR
312.30(b) and (d)(2)).
E. Informed Consent in Trials Involving Long Term Follow-up Observations
Each subject in a clinical investigation must be provided with a description of any
reasonably foreseeable risks from participating in the investigation (21 CFR 50.25(a)(2)).
The informed consent document must describe, among other things, the purposes of the
Contains Nonbinding Recommendations
22
research, the expected duration of the subject's participation and the procedures to be
followed (21 CFR 50.25(a)(1)). Accordingly, the informed consent document must
explain the purpose and duration of LTFU observations, the time intervals, and the
locations at which you plan to request the subjects to have scheduled study visits or be
contacted by other means, and details as to what those contacts will involve (21 CFR
50.25).
When appropriate, the informed consent document must be updated to describe any
adverse reactions that may be associated with the product from your trial or other human
or animal (preclinical) studies (21 CFR 50.25(b)(5)). If the sponsor intends to store blood
or tissue samples for future testing, the informed consent document must convey this
information (21 CFR 50.25(a)(1)). The informed consent should also convey that an
autopsy may be requested to test vector persistence, transgene expression, and related
adverse reactions at the molecular, cellular or tissue level if there are deaths during the
LTFU observation. Sponsors must ensure that investigators submit the informed consent
documents for Institutional Review Board review and approval (21 CFR
312.53(c)(1)(vi)(d)).
We provide additional informed consent recommendations for retroviral vectors in
section V.F.3 of this document.
F. Special Considerations Regarding Integrating Vectors
The recommendations in this section apply exclusively to subjects in clinical trials who
received GT products that are integrating vectors, such as transposon elements,
gammaretroviral, lentiviral, other retroviral vectors, or GT products that are cells modified
ex vivo by integrating vectors or transposon-based vectors. See section VI. for post
licensure considerations. Because of the risk of developing leukemias and premalignant
conditions (clonal cell expansion) due to integration of gammaretroviral vectors and
lentiviral vectors (as described in sections III.B and III.C of this document), we are also
providing additional recommendations (as listed below) for collection of data in studies
in which subjects are exposed to integrating vectors.
1. Data Collection
We recommend that you perform assays to assess the pattern of vector
integration sites in relevant surrogate cells (e.g., determine whether the
dominant clone(s) persist, assess what the relative contribution of the
dominant clone(s) is, and whether the clonal outgrowth observed results in
any (hematologic) malignancies). We consider an assessment of the
vector integration pattern to be relevant in subjects in gene therapy clinical
trials involving integrating vectors when: (1) the target cells are known to
have a high replicative capacity and long survival, and (2) a suitable
surrogate is accessible for assay. For example, hematopoietic stem cells
have a high replicative capacity and long survival; peripheral blood could
serve as a surrogate for testing for vector persistence if hematopoietic stem
Contains Nonbinding Recommendations
23
cells are the target of your gene therapy. In those cases where peripheral
blood is the surrogate, analyses on purified subsets of hematopoietic cells
(e.g., lymphocytes vs. granulocytes) may be performed, if deemed
appropriate to the study. As an alternative example, if the integrating
vector is used for in vivo transduction of liver hepatocytes, you may not
need to perform this analysis, since terminally differentiated hepatocytes
are non-dividing cells under normal circumstances, and there is no
reasonable surrogate that allows for non-invasive testing of vector
persistence. Please refer to the following recommendations for developing
methods and plans for performing these analyses.
a. The choice of method to assess the pattern of vector integration
sites should be based upon data with appropriate positive and
negative controls (i.e., target cells with a known number and sites
of vector copies integrated vs. target cells with no vector
integrants). Studies should be performed to provide information
about the assay sensitivity, specificity, and reproducibility.
b. We recommend that you perform an analysis to assess the pattern
of vector integration sites if at least 1% cells in the surrogate
sample are positive for vector sequences by PCR. As an
alternative, you may base the decision to analyze for clonality of
vector integration sites on an evaluation of the sensitivity of the
assay system used to detect clonality.
c. We recommend that you test for vector sequences by PCR in
subject surrogate samples obtained at intervals of no greater than
six months for the first five years and then no greater than yearly
for the next ten years, or until such time that no vector sequences
are detectable in the surrogate sample.
d. We recommend that you perform an analysis to determine the site
of vector integration if the analysis of a subject’s surrogate cells
suggests a predominant clone (e.g., oligoclonal pattern of vector
insertions) or monoclonality. In addition, if you detect a
predominant integration site, test for persistence by performing
another analysis for clonality no more than three months later.
e. When the nucleotide sequence adjacent to the site of the vector
integration has been determined, we recommend that you compare
the identified integration site sequence with known human
sequences in the human genome database and other databases that
document oncogenes to determine whether the identified
sequences are known to be associated with any human cancers.
f. While we recognize that oligoclonality or even monoclonality
itself will not a priori result in a malignancy (Refs. 15, 31-33), we
also recognize that these changes increase the risk of a malignancy,
and therefore, we recommend that you institute a plan to monitor
the subject closely for signs of malignancy if any of the following
conditions pertain:
Contains Nonbinding Recommendations
24
i. Persistent monoclonality;
ii. Clonal expansion (e.g., the percent cells positive for a
particular vector integration site is shown to increase over
multiple time points); or
iii. Evidence of vector integration near or within a locus
known to have oncogenic activity.
g. To screen for specific disease entities, we recommend that you use
established methods and/or seek advice from clinicians with
expertise in screening for the health care risks to which, according
to your evidence, your subjects may be exposed.
For retroviral (e.g., gammaretroviral and lentiviral) vector-based GT products, additional
follow-up monitoring for the presence of replication competent retrovirus (RCR) may be
necessary. For details regarding duration of the follow-up monitoring for RCR and
methods, please refer to the document “Testing of Retroviral-Based Human Gene
Therapy Product for Replication Competent Retrovirus During Product Manufacture and
Patient Follow-up; Draft Guidance for Industry” dated July 2018.
We recommend that GT products with transposon elements be monitored in a similar
way as gammaretroviral or lentiviral vectors. This recommendation is based on the
potential safety risk of insertional mutagenesis due to the random integration directed by
the transposon, and due to the potential for remobilization of a transposon (secondary
transposition-insertion event) as a result of the continuing presence of the transposase
enzyme in target cells. Yet, if your GT product contains transposon elements you may
propose shorter LTFU observation by providing adequate supporting data/information
related to your product.
2. Data Reporting
If no evidence of oligoclonality or monoclonality is observed, we
recommend that you report a summary of all analyses for the pattern of
vector integration sites in narrative or tabular form in the annual report to
your IND (21 CFR 312.33(b)(5)). However, if evidence of oligoclonality
or monoclonality is observed, you must submit this essential information
in an information amendment to the IND (21 CFR 312.31(a)). We
recommend that you submit this amendment within 30 days of receiving
the report of such an observation.
3. Informed Consent in Trials Involving Retroviral Vectors
Please see section V.E for general consideration of LTFU observation
informed consent. In accordance with 21 CFR 50.25(a)(2), for all clinical
trials in which subjects are exposed to retroviral vectors, the informed
consent documents must include current, complete and accurate disclosure
of the development of leukemias in the clinical trials where such adverse
Contains Nonbinding Recommendations
25
events were reported. Further, the information that is given to the subject
or his/her representative must be in language understandable to the subject
or representative (21 CFR 50.20). We provide the following list as
information and language we recommend be included in the informed
consent document, where applicable, in the section describing the risks
associated with the study agent:
a. Description of study agent - The study involves giving a person
some cells that have been changed by a retroviral vector. A
retroviral vector is a virus that can insert genetic material into cells.
b. Mechanism of action for retroviral vectors - When retroviral
vectors enter a normal cell in the body, the deoxyribonucleic acid
(DNA) of the vector inserts itself into the normal DNA in that cell.
This process is called DNA integration.
c. Effect of DNA integration - Most DNA integration is expected to
cause no harm to the cell or to the patient. However, there is a
chance that DNA integration might result in abnormal activity of
other genes. In most cases, this effect will have no health
consequences. However, in some cases, abnormal activity of a
gene may cause unpredictable harm such as the development of
cancer.
d. Discussion of delayed adverse event, leukemia-like malignancy,
occurring in human studies - It is important that you know about
some cancers that occurred in another gene therapy research study.
Clinical studies were conducted in France and United Kingdom to
treat a disease called X-linked Severe Combined
Immunodeficiency (SCID). Years after receiving cells that were
modified by a retroviral vector, a significant number of the
children in this small study developed a leukemia-like malignant
disease (cancer). One child died from the cancer. A group of
experts in this field studied the results from tests performed on
these children’s blood cells. They concluded that cancer was
caused by the retroviral vector DNA. Still, most of the children
with X-linked SCID who have received experimental gene therapy
have not been found to have cancer at this time. Although they
appear healthy, we still do not know whether they, too, will
develop cancer.
e. Risk of malignancy for this study - We do not know if the
retroviral vector used in this protocol might cause cancer.
However, you should be aware that the DNA contained in
retroviral vectors will integrate into your DNA and that under
some circumstances; this has been known to cause cancer months
to years later.

Contains Nonbinding Recommendations
26
G. Special Considerations Regarding Product Involving Genome Editing
While the general principles for LTFU observations of GT products also apply to LTFU
observations of genome editing products, we recommend that you consider the following:
1. Propose a specific plan to monitor for delayed adverse events based on the
off-target activities noted in your preclinical studies (e.g., in vivo, in vitro
and in silico analysis such as INDEL, (insertion and deletion of bases in a
genome). For example, if the off-target activity involves a tumor
suppression gene in liver cells, you may propose a monitoring plan for
evaluation of occurrence of liver cancer as part of the LTFU observation.
2. Propose a monitoring plan regarding the adverse events from the specific
organ system that the genome editing targets, that may include history and
physical examination, general and specific laboratory tests, and imaging
studies.
3. If direct monitoring of the target tissue is not ethical or feasible, such as,
the brain tissue, you may propose an alternative plan for monitoring of the
product’s effects.
4. Quantitate the relationship between the off-target and on-target activities,
and use the measured level of on-target activity to predict the level of off-
target activity and, if appropriate, establish a follow-up plan;
5. If the genome editing product is delivered via systemic administration,
clinical safety monitoring may be directed not only to off-target activity of
the target organ or tissue, but also to other off-target effects that may occur
in other tissues and organs. Accordingly, consider including appropriate
monitoring tests with a rationale for the proposed monitoring in your
LTFU protocol.
VI. GENERAL CONSIDERATIONS FOR POST-MARKETING MONITORING
PLANS FOR GENE THERAPY PRODUCTS
The number of subjects receiving GT products is typically limited during clinical investigations.
In addition, the recommended LTFU (e.g., 15-year period) will often not elapse for all subjects
who received an investigational GT product in the pre-marketing program before the product is
licensed. Considering that, the safety data generated during clinical trials may not capture all
possible delayed adverse events. Therefore, continuing LTFU observations is often essential
even after a product’s licensure. Consequently, we recommend that at the time of your BLA
submission you submit a Pharmacovigilance Plan (PVP) as described in the FDA Guidance for
Industry; E2E Pharmacovigilance Planning (Ref. 34). Of note, the ongoing or the planned LTFU
study (under an IND) could be a component of the PVP (post-licensure). The contents of PVP
for a particular GT product will depend on its safety profile and will be based on data, which
Contains Nonbinding Recommendations
27
includes the pre-licensure clinical safety database, published literature, and known product-class
effects, among other considerations.
Routine surveillance for licensed biological products includes adverse event (AE) reporting in
accordance with 21 CFR 600.80 (reporting of expedited and non-expedited AEs as well as
periodic safety reports). Submission of reports for serious, life-threatening and unexpected
adverse events may also be required in an expedited manner beyond routine required reporting.
Additional pharmacovigilance elements may be needed, such as those described in the FDA
Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment; Guidance for
Industry dated March 2005 (Ref. 35), for LTFU of patients treated with GT products. For
instance, we may recommend that you establish a registry, or use an existing patient registry, to
systematically capture and track data from treated patients with solicited sample collection (if
applicable), and follow-up of adverse events to resolution or stabilization to collect additional
pertinent data. It may be appropriate to establish a registry system to specifically capture adverse
event data from treated patients who receive a GT product. This registry system can be a part of
the PVP plan and reviewed at the time of licensure.
For any proposed or required post-marketing studies or clinical trials, we recommend that you
include in your BLA submission the study protocol, statistical analysis plan, and a projected
schedule of anticipated study milestones. Your study protocol should include specific adverse
events of interest that you intend to evaluate, and the duration of observation for all patients
enrolled in your post-marketing study. LTFU data collected post-licensure should be submitted
in the BLA annual report (See Appendix 1 of this document).
During our review of your BLA, we will also assess whether a Risk Evaluation and Mitigation
Strategy (REMS) is necessary to ensure that the benefits of your product outweigh its risks. If
you consider that risk mitigation measures are necessary for the safe use of your product, you
may voluntarily submit your proposed REMS as described in Format and Content of a REMS
Document; Draft Guidance for Industry; Drug Safety dated October 2017 (Ref. 36).
If at the time of BLA approval FDA determines it is necessary to initiate LTFU observations,
you should submit the study protocol to your existing IND, with a cross-reference letter to the
BLA submission explaining that the protocol was submitted to the IND.
VII. LONG TERM FOLLOW-UP UNDER SPECIAL CIRCUMSTANCES
A sponsor may cease to operate or may decide to inactivate, transfer or withdraw an IND before
completion of LTFU observations for all subjects exposed to the GT product under its IND.
Under such circumstances, prior to inactivating, transferring or withdrawing an IND, or ceasing
to operate, we recommend that a sponsor consult with OTAT on the plans for completion of
LTFU observation.

Contains Nonbinding Recommendations
28
VIII. DEFINITIONS
The following definitions apply to this guidance:
Engineered site-specific endonucleases: Enzymes that are capable of precisely cleaving
(cutting) DNA based on specific recognition of the DNA sequence at or near the site of DNA
cleavage.
Genome editing: A process by which DNA sequences are added, deleted, or replaced at
specified location(s) in the genome using site-specific nuclease-dependent or nuclease-
independent technologies.
Gene transfer: The transfer of genetic material into a cell.
Human gene therapy: Human gene therapy seeks to modify or manipulate the expression of a
gene or to alter the biological properties of living cells for therapeutic use.
Human gene therapy product: FDA generally considers human gene therapy products to
include all products that mediate their effects by transcription or translation of transferred genetic
material or by specifically altering host (human) genetic sequences. Some examples of gene
therapy products include nucleic acids (e.g., plasmids, in vitro transcribed ribonucleic acid
(RNA)), genetically modified microorganisms (e.g., viruses, bacteria, fungi), engineered site-
specific nucleases used for human genome editing,
10
and ex vivo genetically modified human
cells. Gene therapy products meet the definition of “biological product” in section 351(i) of the
Public Health Service (PHS) Act (42 U.S.C. 262(i)) when such products are applicable to the
prevention, treatment, or cure of a disease or condition of human beings.
11
Integration (of DNA): The process whereby exogenous DNA sequences become incorporated
into a genome.
Latency (of a viral infection): A period of time during which a virus is present in the host
without producing overt clinical symptoms.
Persistence: With respect to transferred or altered genetic material, the continued presence of
transferred or modified genetic sequences in the host after acute exposure to a gene therapy
agent, whether due to integration of the genetic sequence into the host genome, deletion,
insertion, or otherwise modified following genome editing, or to latent infection with the viral
vector bearing the genetic sequence.
Reactivation (of a viral infection): The re-emergence of a symptomatic or asymptomatic viral
infection following a period of latency.
10
Human Genome Editing: Science, Ethics, and Governance. The National Academies Press; 2017.
https://www.nap.edu/read/24623/chapter/1#xvii
.
11
See Federal Register Notice: Application of Current Statutory Authorities to Human Somatic Cell Therapy
Products and Gene Therapy Products (58 FR 53248, October 14, 1993),
https://www.fda.gov/media/76647/download
.
Contains Nonbinding Recommendations
29
Transgene: An exogenous gene that is introduced into a host cell.
Vector sequences: Refers to specific sequences of nucleotides, either DNA or RNA, that have
been introduced into a gene therapy product and includes the vector backbone, transgene(s), and
regulatory elements.
Vector: A vehicle consisting of, or derived from, biological material that is designed to deliver
genetic material. Examples include plasmids, viruses, and bacteria that have been modified to
transfer genetic material.

Contains Nonbinding Recommendations
30
IX. REFERENCES
1. Guidance for Industry: Gene Therapy Clinical Trials – Observing Subjects for Delayed
Adverse Events, November 2006. https://www.fda.gov/media/72225/download
2. Human Genome Editing: Science, Ethics, and Governance, National Academy Press,
Washington D.C., 2017.
3. Donahue, RE, et al., Helper virus induced T cell lymphoma in nonhuman primates after
retroviral mediated gene transfer, Journal of Experimental Medicine, 1992;176:1125-
1135.
4. Hacein-Bey-Abina, S, et al., Sustained correction of X-linked severe combined
immunodeficiency by ex vivo gene therapy, N. Engl. J. Med, 2002; 346:1185-1193.
5. Biological Response Modifiers Advisory Committee (BRMAC), Meeting Minutes,
Department of Health and Human Services, Food and Drug Administration, CBER,
October 10, 2002.
6. Biological Response Modifiers Advisory Committee, Meeting Minutes Department of
Health and Human Services (BRMAC), Food and Drug Administration, CBER,
November 17, 2000; April 6, 2001; and October 24, 2001.
7. Nyberg, K, et al., Workshop on long-term follow-up of participants in human gene
transfer research, Molecular Therapy, 2004; 6:976-980.
8. Cornetta, K, et al., Screening Clinical Cell Products for Replication Competent
Retrovirus: The National Gene Vector Biorepository Experience, Mol. Ther. Methods
Clin. Dev., 2008; 10:371-378.
9. Marcucci, KT, et al., Retrovial and Lentiviral Safety Analysis of Gene-Modified T Cell
Products and Infused HIV and Oncology Patients, Mol. Ther., 2018; 26(1):269-279.
10. Hacein-Bey-Abina, S, et al., LMO2-associated clonal T cell proliferation in two patients
after gene therapy for SCID-X1, Science, 2003; 302:415-419.
11. Hacein-Bey-Abina, S, et al., Insertional oncogenesis in 4 patients after retrovirus-
mediated gene therapy of SCID-X1, J. Clin Invest, 2008; 118:3132-3142.
12. Braun, et al., Gene Therapy for Wiskott-Aldrich Syndrome—Long term Efficacy and
Genotoxicity, Science Translational Medicine, 2014; 6:227.
13. Cavazzana-Calvo, et al., Gene therapy of human severe combined immunodeficiency
(SCID)-X1 disease, Science, 2000; 288:669.
14. Howe, et al., Insertional mutagenesis combined with acquired somatic mutations causes
leukemogenesis following gene therapy of SCID-X1 patients, J Clin Invest, 2008;
118:143-150.
15. Cavazzana-Calvo, et al., Transfusion independence and HMGA2 activation after gene
therapy of human β-thalassaemia, Nature, 2010; 467:318-322.
16. Cavazzana-Calvo, et al., Haematopoietic stem cell transplantation for SCID patients:
where do we stand?, British Journal of Haematology, 2013; 160:146-152.
17. Niedere, HA and CRM Bangham, Integration site and clonal expansion in human chronic
retroviral infection and gene therapy, Viruses, 2014; 6:4140-4164.

Contains Nonbinding Recommendations
31
18. Sakuma, T, et al., Lentiviral vectors: Basic to translational Biochem J, 2012; 443:603-
618.
19. Maetzig, T, et al., Gammaretroviral vectors: Biology, Technology and Application,
Viruses, 2011; 3:677-713.
20. Aronovich, E, et al., The Sleeping Beauty transposon system: a non-viral vector for gene
therapy, Hum Mol Genet, 2011; 20:R14-R20.
21. Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene
Therapy Products, November 2013. https://www.fda.gov/media/87564/download
22. Bauer, S, Current FDA approach for preclinical vector biodistribution studies,
Recombinant DNA Advisory Committee Meeting, March 12, 1999.
23. Shayakhmetov, DM, et al., A high-capacity, capsid-modified hybrid adenovirus/adeno-
associated virus vector for stable transduction of human hematopoietic cells, Journal of
Virology, 2002; 76(3):1135-1143.
24. Goncalves, MA, et al., Stable transduction of large DNA by high-capacity adeno-
associated virus/adenovirus hybrid vectors, Virology, 2004; 321(2):287-296.
25. Picard-Maureau, M, et al., Foamy virus—adenovirus hybrid vectors, Gene Therapy,
2004; 11(8):722-728.
26. Yant, SR, et al., Transposition from a gutless adeno-transposon vector stabilizes
transgene expression in vivo, Nature Biotechnology, 2002; 20(10):999-1005.
27. Wang, Z, et al., Detection of integration of plasmid DNA into host genomic DNA
following intramuscular injection and electroporation, Gene Therapy, 2004; 11(8):711-
721.
28. Guidance for Industry: Considerations for the Design of Early-Phase Clinical Trials of
Cellular and Gene Therapy Products, August 2015.
https://www.fda.gov/media/106369/download
29. E6 (R2) Good Clinical Practice: Integrated Addendum to ICH E6 (R1): Guidance for
Industry, March 2018. https://www.fda.gov/media/93884/download
30. E2F Development Safety Update Report; Guidance for Industry, August 2011.
https://www.fda.gov/media/71255/download
31. Ott, MG, et al., Correction of X-linked chronic granulomatous disease by gene therapy,
augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1, Nature
Medicine, 2006; 12(4):401-409.
32. Fraietta, JA, et al., Disruption of TET2 promotes the therapeutic efficacy of CD19-
targeted T cells, Nature, 2018; 558(7709):307-312.
33. Schmidt, M, et al., Clonality analysis after retroviral-mediated gene transfer to CD34+
cells from the cord blood of ADA-deficient SCID neonates, Nature Medicine, 2003;
9(4):463-468.
34. E2E Pharmacovigilance Planning; Guidance for Industry, April
2005. https://www.fda.gov/media/71238/download

Contains Nonbinding Recommendations
32
35. Guidance for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic
Assessment, March 2005. https://www.fda.gov/media/71546/download
36. Format and Content of a REMS Document; Draft Guidance for Industry, October 2017.*
https://www.fda.gov/media/77846/download
*When finalized, this guidance will represent FDA’s current thinking on this topic.
Contains Nonbinding Recommendations
33
APPENDICES
APPENDIX 1: INFORMATION FOR LONG TERM FOLLOW-UP (LTFU)
OBSERVATION ANNUAL REPORT
Category Required LTFU Data Rationale
Protocol Title
“Long Term Follow-Up Observation Annual
Report”
The placement of this title will
facilitate FDA to search for
LTFU data in our database
LTFU Protocol Status
Total length (years)
Starting date
Total number of subjects enrolled
Subjects that have completed LTFU observation
Remaining subjects on LTFU observation
This will serve as a brief
summary.
Product Information
Vector persistence
Clonality analyses
RCR
On and off-target analyses for products that involve
genome editing
This is the focus of the product
safety assessment in the LTFU
protocol and provides important
information for monitoring, and
for determination of the length of
the LTFU observation.
Preclinical
Information
New preclinical data
Relevant findings from the literature
This provides data and signals to
guide the direction of LTFU
observation.
Clinical Information
Any related delayed adverse event with brief
narrative
Oncological, neurological, hematological, auto-
immune or other disorder
Causal analyses based on evidence from clinical,
laboratory, molecular, cytogenetic, histological,
HLA analysis, deep sequencing data
Serious adverse events
Evidence for persistence of the product/therapeutic
protein/sequences, and durability of the clinical
effects
This is the focus of the product
safety assessment in LTFU
observation, and serves as a
guide for the types of AE, organ
systems, and methodology to
attribute AE/Serious Adverse
Event (SAE) to the GT product.
The durability of clinical effect
also allows for an assessment of
product efficacy in the LTFU
observation report, but inclusion
of such data is at the sponsor’s
discretion.
Revision of LTFU
protocol
Rationale for modifying LTFU observation
FDA agreement to revised LTFU protocol: synopsis
of meeting(s) discussion/email communication
Discussion and date of discontinuation
This will provide an opportunity
for revising the content and
length of the LTFU observation
based on data collected in the
studies or other relevant
information.
Contains Nonbinding Recommendations
34
APPENDIX 2: SAMPLE TEMPLATE: LONG TERM FOLLOW-UP (LTFU)
OBSERVATION ANNUAL REPORT
Category
List of LTFU data
Annual reporting
Protocol title
“Long Term Follow-Up
Observation Annual Report”
[product name]: LTFU2017
annual report for protocol [#]
LTFU protocol
status
Total length (years):
15 years
Starting date:
October 30, 2009
Total number of subjects
enrolled:
30
Subjects that have completed
LTFU observation:
0
Remaining subjects on LTFU
observation:
20 (2 deaths, 5 lost to follow-up,
3 drop outs)
Product
information
Vector persistence:
PCR
1
of [name] transgene
positive in 17 of 20 subjects still
on study at 5 yrs and 3 subjects
at 7 yrs.
Clonality analyses:
No clones more than 1% for
more than 1 testing period
RCR
ND
2
, request to discontinue RCR
testing
On and off-target analyses for
products that involve genome
editing
NA
3
Preclinical
information
New preclinical data
Final study report for large
reproductive toxicity study in
normal SD rats (study report
[#]). Published in [journal
citation].
No additional studies ongoing at
this time.
Relevant findings from the
literature
No new literature on [x] disease
at this time.
Clinical
information
Any related delayed adverse
event with brief narrative
One case of rash that resolved
with steroids. No other
symptoms. PCR of rash biopsy
was negative for vector.
Oncological, neurological,
hematological, auto-immune or
other disorder
Secondary tumor on left ear,
negative for vector sequences by
PCR. Unrelated, melanoma.
Causal analyses based on
evidence from clinical,
NA
Contains Nonbinding Recommendations
35
laboratory, molecular,
cytogenetic, histological, HLA
analysis, deep sequencing data
Serious adverse events
2 deaths due to sepsis, related to
underlying disease.
No other unexpected SAE
reported
Evidence for persistence of the
product/therapeutic
protein/sequences, and durability
of the clinical effects
20 subjects are still on study with
vector persists in BM and PBMC
samples, and clinical benefit
observed. All twenty subjects
have reconstituted immune
system, with some b cell aphasia
and low platelet counts in three
subjects, however no
transfusions needed to date.
Revision of LTFU
Protocol
Rationale for modifying LTFU
observation
All RCR testing results negative
(n=150 samples). Risk
assessment determined very low
risk of RCR developing in
subjects at this time.
FDA agreement to revised LTFU
protocol: synopsis of meeting(s)
discussion/email communication
Revision to LTFU discussed
during pre-BLA meeting [date].
RCR testing will no longer
performed for LTFU protocol [#]
Discussion and date of
discontinuation
NA
1
polymerase chain reaction
2
none detected (ND)
3
not applicable (NA)
